

CAPA – Corrective and Preventive Action
A Corrective and Preventive Action (CAPA) procedure is a documented process within Quality Management Systems (QMS) that outlines how corrective and preventive actions are managed. It is designed to identify, analyse, and resolve recorded issues by systematically investigating root causes and implementing corrective and preventive measures.
The purpose of the CAPA procedure is to ensure regulatory alignment, product integrity, and operational efficiency by addressing quality issues at their source. Effective corrective actions (CA) eliminate defects, inconsistencies, and process failures. Preventive actions (PA) proactively address potential risks, preventing issues from arising.
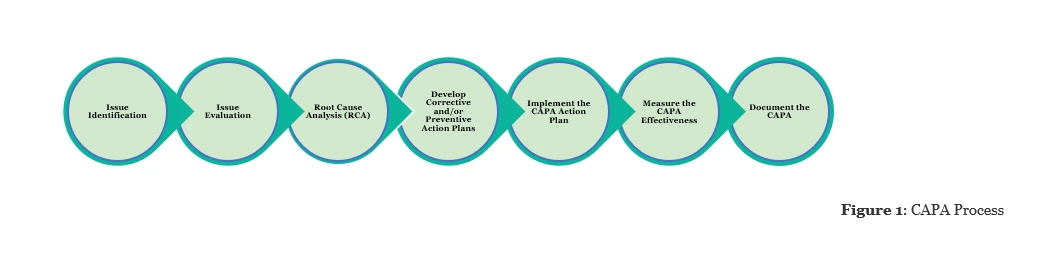
CAPA PROCESS:
1. Issue Identification
The CAPA process starts with clearly articulating the problem, deviation, or non-conformance, including the source, such as customer complaints, audits, or production issues, and documenting the supporting evidence.
2. Issue Evaluation
After identifying the issue's severity, frequency, and impact, they need to be understood to determine its priority. The issues then need to be categorised by the attention required, with high-risk situations prioritised.
3. Root Cause Analysis
It is the critical step in CAPA analysis that helps to determine the weakness and underlying factors of risk. Tools such as Fishbone diagrams, 5 Whys, and Fault Tree Analysis are used.
4. Develop Corrective and/or Preventive Action Plans
Based on the RCA, create a corrective action (CA) plan to address the issue or a preventive action (PA) plan to mitigate future potential, ensuring the plans target the root cause, not just symptoms.
5. Implement the CAPA Action Plan
After CA/PA plan development, carry them out within the set timelines by assigning responsibilities, allocating resources, and communicating plans to relevant stakeholders.
6. Measure the CAPA Effectiveness
After the plans are implemented, their effectiveness needs to be monitored to ensure that the identified issues have been successfully addressed and future risks have been mitigated.
7. Document the CAPA
Detailed records of all activities, including investigation reports, action plans, implementation updates, and effectiveness checks, need to be maintained.
Corrective Action Plan vs Preventive Action Plan
| Corrective Action | Preventive Action |
|---|---|
| Reactive approach | Proactive approach |
| Action taken to address the root cause and prevent recurrence. | Action taken on potential causes of failures to prevent their occurrence. |
| Deals with a nonconformity that has occurred. | Deals with potential nonconformity; no failure has yet occurred. |
| Requires root cause analysis | Requires risk or trend analysis |
| Documentation acts as evidence that the problem was recognised, addressed, and controlled to prevent recurrence. | Documentation shows a system capable of anticipating, identifying, and preventing potential issues. |
Requirements for an Effective CAPA :
8D approach for problem resolution
The 8D methodology is a structured approach to addressing and resolving problems. Its primary goals are to identify root causes, implement corrective actions, and resolve issues through teamwork, thereby enhancing the product and processes.
Assembling of a Knowledgeable Team
To create the team, designate a leader and select members with relevant expertise for the specific issue. Make sure the team is cross-functional, with members who deeply understand the product or process involved. Clearly outline the team's goals and objectives to coordinate efforts and steer the problem-solving process efficiently.
Clear Definition of the Problem Using the 5W2H Method
The problem should be clearly identified using the 5W2H (what, when, where, who, which, how) method along with a process flow diagram (PFD). Make sure the problem definition is based on facts, not opinions.
Development and Validation of Long-Term Solutions
Identify and implement permanent corrective actions (PCAs) that effectively address the root cause of the problem. These actions should resolve the issue for the customer. Select the most suitable solution from all options, and document and verify the PCA to ensure its effectiveness.
Adjustment of Systems to Prevent Recurrence
Ensure CA are consistently applied to similar machines, products, and services to avoid recurring issues. Update related systems, processes, procedures, and documents, including control plans and FMEAs, to prevent recurrence. Regularly review the effectiveness of these permanent CA to ensure ongoing success.
Why is CAPA necessary?
• The ICH Q10 Guideline from the International Council for Harmonisation establishes a Pharmaceutical Quality System (PQS) that guarantees uniform product quality by implementing robust risk management. According to Section 3.2.2, CAPA mandates that organisations identify, investigate, and address quality issues through corrective and preventive actions, coupled with ongoing monitoring to avoid future problems.
• FDA 21 CFR Part 820 outlines the Quality System Regulation (QSR) for medical device manufacturers in the U.S. It specifies that CAPA (Corrective and Preventive Actions), as detailed in Subpart J, Section 820.100, requires identifying, documenting, and investigating nonconformities, as well as implementing corrective and preventive measures to promote ongoing improvement.
• FDA 21 CFR Part 211 is a U.S. regulation that specifies current Good Manufacturing Practices (cGMP) for pharmaceuticals to ensure quality and safety. In this context, CAPA (Subpart J, Section 211.192) emphasises documenting nonconformities, investigating root causes, and validating processes to stay compliant.
• ISO 13485:2016 is an international standard governing medical device quality management system. Clauses 8.5.2 and 8.5.3 on CAPA mandate organisations to implement early warning systems and feedback channels, monitor product quality and undertake risk-based corrective actions, analyse data, and inform regulators about adverse events to maintain compliance.
• EU GMP guidelines establish standards for pharmaceutical manufacturing quality to ensure product safety and efficacy. According to EU GMP (Chapter 1, Section 1.4.xi and Chapter 8, Sections 8.16 to 8.19), CAPA requires organizations to regularly document and review preventive actions, conduct root cause analyses to enhance product safety, and optimize manufacturing processes to sustain ongoing regulatory compliance.
• WHO GMP establishes international standards for the manufacturing of pharmaceutical and healthcare products. According to Annex 3, Section 14.2, the CAPA in the WHO GMP mandates that companies establish clear procedures, perform root cause analyses, carry out corrective and preventive actions, and evaluate their effectiveness to maintain high-quality standards.
• The European Union Medical Device Regulation (EU MDR) sets requirements to ensure the safety and effectiveness of medical devices available in the EU. According to the MDR (Chapter I, Article 2(68)), CAPA focuses on identifying and addressing nonconformities to keep products safe and compliant with regulatory standards.
• The European Union In Vitro Diagnostic Regulation (EU IVDR) sets standards for the safety, performance, and regulatory approval of in vitro diagnostic medical devices in the EU. According to IVDR (Chapter I, Article 2(70)), CAPA involves identifying, investigating, and resolving nonconformities through actions aimed at eliminating their root causes or other issues.
• ISO 9001:2015 is an international standard for Quality Management Systems (QMS) that applies across various industries to maintain consistent product and service quality. Clause 10.2 on CAPA highlights the importance of correcting nonconformities and preventing recurrence using risk-based methods.
CAPA Process Flow
Issue Identification → Issue Evaluation → Root Cause Analysis (RCA) → Develop Corrective and/or Preventive Action Plans → Implement the CAPA Action Plan → Measure the CAPA Effectiveness → Document the CAPA